Analisi gascromatografica degli
acidi grassi volatili prodotti durante la digestione anaerobica
Gascromatographic determination of volatile fatty acids produced by
anaerobic digestion
Fabrizio Malaspina*, Carmela M. Cellamare*, Loredana Stante*, Giuseppe Bortone*
e Andrea Tilche**
*ENEA, Sezione Depurazione e Ciclo
dell'Acqua, Via Martiri di Monte Sole,
4-40129, Bologna Italia
**European Commission Research D G -Direction D. I. Unit Head- Key
Action"Sustainable Management and Quality of Water" Bureau: Square de
Meeus 8, 7/49, 1050 Bruxelles-Belgium
Abstract
La digestione anaerobica porta alla formazione, come intermedi del processo, di acidi organici a catena corta (AGV). In particolari condizioni, come durante il trattamento di reflui molto zuccherini, tale componente acida può portare ad una eccessiva riduzione del pH fino alla compromissione del processo. La misura della concentrazione degli AGV è un ottimo strumento per prevenire cali di pH, tuttavia occorre che la metodica di analisi sia semplice, rapida e che richieda una preparazione minima del campione, ma che allo stesso tempo sia sufficientemente precisa ed accurata nei dati forniti. Durante una ricerca sulla digestione anaerobica del siero di latte, è stata messa a punto una metodica di determinazione gascromatografica degli AGV che presenta caratteristiche di semplice preparazione del campione, breve durata, ottimi valori (per il controllo del processo) di precisione ed accuratezza, stabilità nel tempo dei risultati analitici e una buona risoluzione e selettività dei singoli acidi volatili.
******************
The anaerobic digestion process carries to the
formation of short chain organic acids (VFA), as intermediate metabolites. This
occurs mainly during the treatment of high sugars contents wastewaters. VFA
could lead to a strong pH reduction with a failure of the anaerobic process.
The measures of the VFA concentration appear an excellent tool in order to
prevent pH drops. The VFA analytical methods should be simple, rapid, with a
minimum sample preparation, but it should be at the same time enough precise
and accurate. During a research on the anaerobic digestion of milk whey, it was
set up a gaschromatographic methods for VFA determination that take short time
and a minimum sample preparation. The methods was checked for main validation
parameters and it given good performances in terms of precision, accuracy,
resolution and selectivity.
******************
Premessa
Durante il processo di digestione anaerobica, e in particolar modo nel trattamento di reflui ricchi di sostanza organica facilmente biodegradabili, il pH appare un fattore critico per l’ottimizzazione ed il controllo del processo stesso (Kodukula et al., 1988; Clark and Speece, 1970). La determinazione degli acidi grassi volatili a catena corta può rappresentare un’importante strumento per controllo del pH, e quindi per la gestione del processo (Jeris and Kugelman, 1973, Ahring et. al. 1995; Pind et al. 1999).
Tra le diverse tecniche analitiche per la quantificazione degli acidi grassi volatili a catena corta (A A.V.V., 1995), quella gascromatografica permette la separazione e l’identificazione dei singoli composti.
Nell'ambito di una ricerca sulla digestione anaerobica del siero di latte (Malaspina et al. 1995, Malaspina et al. 1996) in cui la fase di acidificazione rappresenta uno dei passaggi critici nel processo, è stata messa a punto una procedura semplice di analisi gascromatografica degli acidi grassi volatili prodotti che non prevede la fase di estrazione. Tale metodo presenta il vantaggio della semplicita' della fase di prettratamento del campione; quest’ultimo, infatti, prelevato dal digestore anaerobico veniva centrifugato a 10.000 g; il surnatante veniva filtrato con filtri 0,45 mm, eventualmente diluito, ed iniettato direttamente in colonna.
E' stata valutata la qualità del metodo attraverso la determinazione dell'accuratezza e della precisione, nonché di altri indici secondari, ma comunque utili.
Importanza degli acidi grassi volatili nel processo di digestione anaerobica
La digestione anaerobica è un processo complesso di trasformazione della sostanza organica, operato da diverse popolazioni batteriche, con la produzione di un gas (biogas) costituito prevalentemente da metano e da anidride carbonica. Il processo avviene attraverso diverse reazioni degradative operate da differenti popolazioni microbiche. Durante queste fasi di trasformazione della sostanza organica vengono prodotti acidi organici a catena corta. Questi rappresentano il punto di incrocio delle varie vie metaboliche e quindi rappresentano un indice dell’evoluzione dell’intero processo di degradazione della sostanza organica (Zehnder, 1988). In normali condizioni di processo, la concentrazione degli acidi grassi volatili rimane relativamente stabile, in quanto la produzione di nuovi acidi grassi ad opera dei batteri fermentativi viene compensata dal consumo degli stessi da parte dei batteri acetogeni e metanigeni; mentre in condizioni anomale di processo, come sovraccarichi, presenza di sostanze tossiche, abbassamenti di pH o di temperatura, si può verificare uno squilibrio tra produzione e consumo degli acidi grassi volatili. In questa condizione i batteri acetogeni e metanigeni, molto più sensibili alle variazioni di pH rispetto ai batteri fermentativi, non sono più in grado di fronteggiare la continua produzione di acidi grassi che si accumulano nel mezzo, in particolare quelli a catena lunga, provocando un abbassamento del pH (Zehnder, 1988).
Durante il processo anaerobico è importante quindi, attraverso un costante ed accurato controllo degli acidi grassi volatili, prevedere e quindi prevenire eventuali abbassamenti del pH, che potrebbero compromettere l’intero processo (Rozzi, 1984).
Materiali e metodi
L'analisi gascromatografica
Gli acidi grassi volatili che sono stati rilevati dall'analisi sono quelli di maggior importanza nel processo anaerobico. Essi sono compresi nell'intervallo C2-C7:
Acido acetico Acido Etanoico
Acido propionico Acido Propanoico
Acido n-butirrico Acido Butanoico
Acido i-butirrico Acido Metil-propanoico
Acido n-valerico Acido Pentanoico
Acido i-valerico Acido Metil-butanoico
Acido n-caproico Acido Esanoico
Acido i-caproico Acido Metil-pentanoico
Acido eptanoico Acido Eptanoico
La determinazione degli acidi grassi volatili è stata effettuata tramite Gas Cromatografo DANI 8510 dotato di rivelatore ad ionizzazione di fiamma (FID). Per la gestione dello strumento e dei dati veniva utilizzato un Personal Computer con programma di acquisizione ed elaborazione del cromatogramma “Turbocrom” (Perkin-Elmer Nelson).
Scelta delle condizioni operative
La scelta dei parametri di esercizio rappresenta il risultato di numerose prove effettuate a diverse condizioni allo scopo di rendere il metodo semplice, ma nello stesso tempo adeguato alle esigenze operative della gestione del processo anaerobico.
La scelta della colonna e delle condizioni di esercizio (temperatura, flusso del carrier, etc.) sono state fatte sia sulla base delle indicazioni fornite dai produttori, sia sull'esperienza.
Come metodo di calibrazione è stato scelto quello della standardizzazione interna, che rispetto ad altri metodi, permette di ridurre l’errore causato da piccole variazioni di volume dell’iniezione.
Per la scelta dello standard interno è stata utilizzata una sostanza dotata, alle condizioni di prova, di un tempo di ritenzione intermedio e di una buona risoluzione rispetto ai componenti da eluire e che non fosse presente nel campione da analizzare. Sono state provate numerose sostanze che presentavano tali requisiti, ed è stato scelto l'acido 2,2-dimetilbutirrico.
Dalle prove effettuate sugli standard analizzati, è stato possibile ottenere le condizioni operative riportate di seguito:
Condizioni strumentali
Colonna usata: Alltech
Superox-FA BP, 25 m per 0.53 mm
(Polyethylene
Glicol Ester), Bonded FS OT
Precolonna usata: Polyethylene
Glicol Ester, 0,5 m per 0.53 mm
Carrier Gas: Idrogeno
Temperatura iniettore: 200°C.
Temperatura detector: 220°C.
Temperatura forno: programma
100° per 1 min
rampa
di 5°/min fino a 135°
135°
per 1 min
Durata analisi: 9
min
Pressione carrier: 1.6
Bar
Detector Pressione
aria 0.8 Bar
Pressione
idrogeno 0.4 Bar
Flusso carrier in colonna: 8
ml/min
Flusso idrogeno alla fiamma: 20
ml/min
Volume di iniezione 0,02
µl
Tipo di iniezione: on-column
Metodo di integrazione: Standardizzazione Interna
Metodo di calibrazione e preparazione degli standard
Per effettuare la calibrazione e la sperimentazione è stata utilizzata una miscela di acidi grassi volatili puri (purezza tipo “standard grade”) in soluzione (Supelco). Un ml di tale soluzione conteneva le seguenti quantità di acidi:
- formico 460 ml
- acetico 601
ml
- propionico 741
ml
- n-butirrico 881
ml
- i-butirrico 881
ml
- n-valerico 1022
ml
- i-valerico 1022
ml
- n-caproico 1162
ml
- i-caproico 1162
ml
- eptanoico 1302
ml
Dalla soluzione concentrata sono state preparate le soluzioni standard utilizzate per la calibrazione ed il controllo. Sono stati scelti tre livelli di calibrazione (1, 2, 3) aventi rapporti di concentrazione per i singoli analiti pari a 1:2:4.
Di seguito si riporta il
metodo di preparazione degli standard.
Soluzioni 1 2 3
standard 100ml 200ml 400ml
acqua deionizzata 900ml 800ml 600ml
standard interno 25ml 25ml 25ml
acido ortofosforico 25ml 25ml 25ml
L’intervallo di calibrazione è stato scelto in funzione delle esigenze di controllo del processo. La concentrazione minima di calibrazione è stata, per tutti gli acidi considerati, superiore alla sensibilità analitica del metodo, intesa come il doppio dell’oscillazione più ampia della linea di base cromatografica. I valori delle concentrazioni di sensibilità, sono riportate di seguito:
Acido
grasso Concentrazione
minima
volatile determinabile
(mg/l)
Ac. acetico 47,8
Ac. propionico 35,7
Ac. n-butirrico 27,9
Ac. i-butirrico 29,2
Ac. n-valerico 26,3
Ac. i-valerico 28,9
Ac. n-caproico 29,1
Ac. i-caproico 27,8
Ac eptanoico 27,8
Lo standard interno era costituito da una soluzione di Acido 2-2-Dimetilbutirrico alla concentrazione di 4000 mg/l.
L'aggiunta di acido ortofosforico serviva ad abbassare il pH e mantenere gli acidi volatili in forma indissociata e quindi favorirne la volatilizzazione.
Le concentrazioni (in mg/l) ottenute per ogni acido al relativo livello di calibrazione sono schematizzate di seguito.
Livello 1 2 3
Ac. acetico 60,1 120,2 240,4
Ac. propionico 74,1 148,2 296,4
Ac. n-butirrico 88,1 176,2 352,4
Ac. i-butirrico 88,1 176,2 352,4
Ac. n-valerico 102,2 204,4 408,8
Ac. i-valerico 102,2 204,4 408,8
Ac. n-caproico 116,2 232,4 468,8
Ac. i-caproico 116,2 232,4 468,8
Ac eptanoico 130,2 260,4 520,8
L'analisi veniva eseguita iniettando 0,02 ml di campione preventivamente agitato, direttamente su iniettore per colonne widebore con una microsiringa (Hamilton da 1 ml).
Essendo gli acidi grassi a catena corta molto volatili, soprattutto dopo acidificazione del campione, durante la preparazione e la manipolazione degli standard sono stati seguiti tutti gli accorgimenti richiesti per ridurre la perdita di analiti.
Verifica della qualità dei risultati analitici ottenuti
Per controllare la
qualità dell’analisi gascromatografica degli acidi grassi volatili, sono state
preparate alcune soluzioni standard, (1, 2, 3), ed effettuate 10 analisi per
ogni punto di calibrazione e sono stati messi a confronto i risultati delle varie
ripetizioni. I dati di ciascun acido rilevato ad ogni livello di calibrazione,
sono stati tabulati ed analizzati ottenendo così 27 tabelle, ciascuna con i
risultati di 10 analisi.
L’attendibilità di un risultato è la rispondenza fra il risultato sperimentale ed il valore vero; esso è condizionato da molti fattori, alcuni dei quali a loro volta dipendono dal metodo, altri dall’esecuzione e dall’efficienza della strumentazione (Bottari e Liberti, 1983; AAVV, 1995). I parametri considerati per il controllo della qualità dei dati sono stati l’accuratezza, la precisione, la ripetibilità, la riproducibilità e la stabilità del fattore di risposta dei quali si riporta, di seguito una sintetica definizione.
-Precisione; rappresenta il grado di ripetibilità tra i risultati, è quindi un indice del grado di dispersione dei risultati analitici. Viene espresso tramite la deviazione standard dalla media dei risultati oppure con il suo valore relativo alla media (coefficiente di variabilità).
-Accuratezza : rappresenta un indice dell’esattezza analitica e ci fornisce una misura sulla dispersione dei valori osservati rispetto al valore atteso. L’accuratezza viene solitamente espressa tramite lo scarto medio dei risultati rispetto al valore atteso, ed è indicata come frazione di recupero dell’analisi rispetto al valore noto dell’analita.
-Ripetibilità e Riproducibilità : la ripetibilità rappresenta la precisione riferita ad un solo operatore, mentre la riproducibilità esprime la precisione riferita a più operatori. Lo stesso concetto può essere esteso all’impiego dello stesso strumento oppure a strumenti diversi operanti alle stesse condizioni. Un metodo riproducibile è sempre ripetibile, mentre non è necessariamente vero il contrario. Quindi la riproducibilità esprime il grado di dispersione dei dati analitici dovuti all’operatore (e/o alla strumentazione).
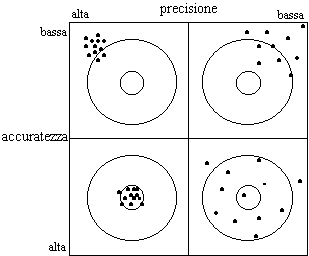
Fig. 2. Rappresentazione grafica dei concetti di precisione e accuratezza
-Fattore di risposta (Fr): è il rapporto delle concentrazioni relative, all’area dei picchi, tra standard interno e analita.
Fr = ![]()
Il Fattore di risposta, fornisce una misura della ripetibilità con cui lo strumento risponde allo stesso livello di standard, permettendo di confrontare e verificare anche letture apparentemente diverse.
Altri parametri considerati per il controllo qualitativo del metodo sono stati la selettività, la risoluzione dei picchi e il fattore di capacità. Tali parametri, di cui si riporta di seguito una definizione sintetica, sono solitamente utilizzati per valutare l’efficienza della colonna, ma forniscono indicazioni utili anche sulla validità della scelta delle condizioni operative quali flusso e temperature.
-Selettività: è la capacità di risolvere i singoli componenti dell’analisi e può essere espressa dal rapporto tra i tempi di ritenzione corretti, cioè detratti del tempo morto del sistema, di due picchi consecutivi. Tale valore, calcolato convenzionalmente tra un picco e quello precedente, deve essere necessariamente maggiore di uno. Sono ritenuti buoni valori compresi tra 1.1 e 1.5.
- Fattore di capacità: rappresenta il tempo di ritenzione del singolo componente relativo al tempo morto del sistema e rappresenta un sistema per indicare in maniera confrontabile i tempi di ritenzione per sistemi diversi.
-Risoluzione: è un indice del grado di separazione tra due picchi adiacenti e viene espresso come rapporto tra la differenza dei tempi di ritenzione dei due componenti e la semisomma della base dei picchi. Tale parametro viene solitamente calcolato per i picchi molto vicini tra loro, dove può essere dubbia la separazione. In genere valori uguali o superiori ad 1.5 indicano la completa separazione dei picchi, ma valori inferiori possono essere ancora accettabili per picchi di dimensioni simili, purché vicini o maggiori a uno, che indica una separazione tra i picchi del 98% (Mentasti e Saini, 1990).
Risultati
I risultati ottenuti sono riportati in maniera sintetica dai grafici che seguono.
Osservando i dati ottenuti riportati nei grafici, possono essere fatte alcune considerazioni sulla precisione, sull’accuratezza (fig. 3-11) e sulla stabilità del fattore di risposta, espresso come coefficiente di variabilità, sia tra i diversi livelli sia tra le analisi dello stesso livello (fig. 11-19). Tali valori devono essere più bassi possibile, come indice di elevata precisione ed accuratezza, perché rappresentano lo scarto relativo dal valore cercato.
Per quanto riguarda i grafici relativi alla precisione ed all’accuratezza, si può osservare che i valori di precisione, fatta eccezione per gli acidi N-caproico ed eptanoico, risultano compresi tra il 4 e 8 %, con i valori più bassi in corrispondenza del secondo livello d’analisi. I valori sono ben al di sotto del 15-16%, percentuali ritenute accettabili per analisi con concentrazione dell’ordine del centinaio di mg·l-1 (Johnson & Stevenson, 1978; Calaresu, 1994).
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig.3-11
Andamento dei valori di accuratezza e precisione percentuale per i singoli
acidi grassi volatili rilevati ai vari livelli analitici. |
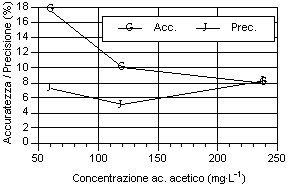
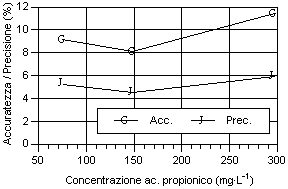
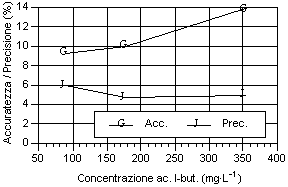
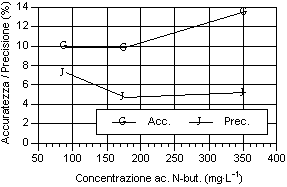
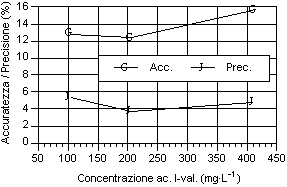
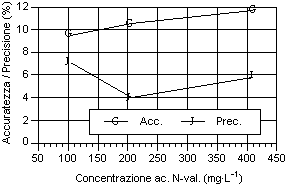
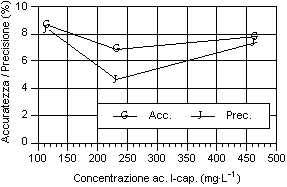
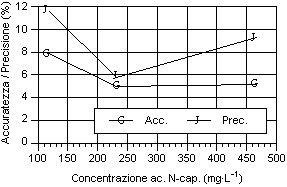
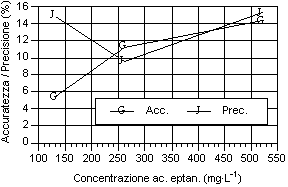
Al contrario l’accuratezza ha mostrato valori relativamente alti, compresi tra l' 8 e il 14 %, con punte fino al 16% per l’acido I-valerico e al 18% per l’acido acetico. In particolare l’acido acetico ha mostrato bassi livelli di accuratezza, sopratutto per il primo livello che aveva picchi relativamente modesti, a causa dell’interferenza nell’analisi da parte di picchi non identificati ed alla vicinanza con il picco di pressione dovuto all’iniezione.
Comunque i valori di accuratezza misurati, rientrano nei limiti di accettabilità per le esigenze di controllo del processo di digestione anaerobica, dove il valore assoluto della concentrazione degli acidi grassi è importante, ma secondario rispetto alla sua variazione nel tempo.
Osservando l’andamento generale si può notare che, salvo alcune eccezioni, si è avuto un calo di accuratezza all’aumentare del livello d’analisi.
Per quanto riguarda la precisione, all'aumentare della concentrazione dell’analita, si è avuto una diminuzione della precisione sia per i singoli acidi sia per i diversi livelli considerati. Questo è osservabile considerando la curva della deviazione standard.
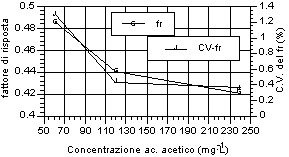
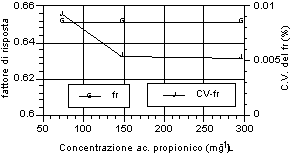
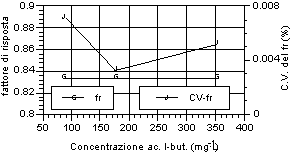
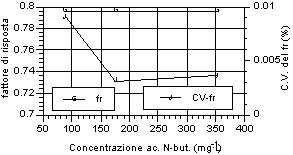
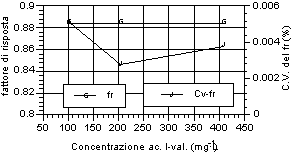
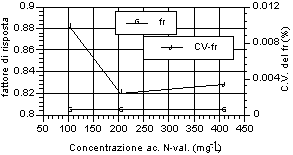
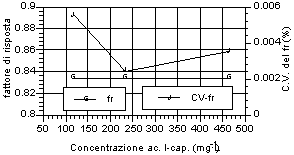
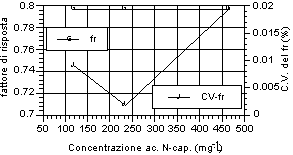
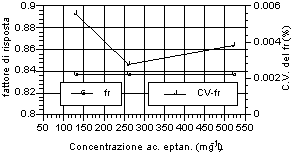
Di seguito (fig. 12-20) vengono riportati i grafici relativi all’andamento del fattore di risposta e del relativo coefficiente di variabilità, indice della stabilità dello stesso. Si può notare che tutti gli acidi hanno presentato una notevole stabilità del fattore di risposta per i tre livelli d’analisi. Anche nell’ambito dei singoli livelli di analisi, il fattore di risposta ha mostrato variazioni minime, come evidenziato dai relativi valori dei coefficienti di variabilità, al di sotto di un fattore 100 al punto percentuale. Tale andamento sta ad indicare una stretta corrispondenza tra il valore dell’area del picco dell’analita, messo in relazione a quella dello standard interno, ed il valore di concentrazione riportato e rappresenta inoltre un ulteriore indice di ripetibilità delle analisi.
A questi risultati ha fatto eccezione la misura relativa all’acido acetico, che ha riportato sia variazioni del fattore di risposta tra i diversi livelli, in particolare per il primo rispetto agli altri due, sia rispetto alle analisi effettuate nell’ambito dello stesso livello, in particolare per il primo, i cui valori del coefficiente di variabilità hanno superato il punto percentuale. Tale situazione è stata probabilmente imputabile alle forti interferenze, avutesi in particolar modo al piu' basso livello di analisi, da parte di picchi non identificati vicini all’analita. Tuttavia anche in questo caso lo scarto relativo, in termini assoluti, rimane molto basso e più che adeguato alle esigenze analitiche relative al controllo di processo.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 12-20. Fattori di risposta ottenuti per i singoli analiti ai vari livelli d’analisi e relativi coefficienti di variabilità per ogni livello analitico. |
La sintesi dei valori di accuratezza e precisione ottenuti durante le prove sono riportate di seguito (Tab.1)
Tab. 1. Precisione ed accuratezza percentuale ottenuti durante le prove.
|
acido |
livello |
1 |
2 |
3 |
|
acetico |
precisione |
7.29 |
5.05 |
8.25 |
|
|
accuratezza |
17.68 |
10.00 |
7.97 |
|
propionico |
precisione |
5.31 |
4.55 |
5.94 |
|
|
accuratezza |
11.34 |
10.04 |
14.06 |
|
I-butirrico |
precisione |
6.01 |
4.77 |
5.00 |
|
|
accuratezza |
13.66 |
14.79 |
20.28 |
|
N-butirrico |
precisione |
7.28 |
4.74 |
5.15 |
|
|
accuratezza |
14.53 |
14.44 |
19.94 |
|
I-valerico |
precisione |
5.33 |
3.64 |
4.79 |
|
|
accuratezza |
19.37 |
18.74 |
22.96 |
|
N-valerico |
precisione |
7.11 |
3.96 |
5.86 |
|
|
accuratezza |
16.24 |
17.95 |
19.93 |
|
I-caproico |
precisione |
8.31 |
4.63 |
7.35 |
|
|
accuratezza |
16.71 |
13.25 |
15.06 |
|
N-caproico |
precisione |
11.68 |
5.80 |
9.25 |
|
|
accuratezza |
15.22 |
9.61 |
10.00 |
|
eptanoico |
precisione |
14.90 |
9.59 |
15.24 |
|
|
accuratezza |
11.76 |
24.45 |
31.04 |
Tab. 2. Risoluzione e selettività degli analiti durante le prove.
|
acido |
tempo rit. corr. (min) |
selettività |
fatt. di capacità |
risoluzione |
|
acetico |
1.37 |
1.48 |
3.42 |
3.4-3.9 |
|
propionico |
2.03 |
1.12 |
5.07 |
0.95-1.16 |
|
I–butirrico |
2.28 |
1.28 |
5.7 |
2.44-2.98 |
|
N-butirrico |
2.92 |
1.15 |
7.3 |
1.75-2.14 |
|
I-valerico |
3.38 |
1.26 |
8.45 |
3.15-4.2 |
|
N-valerico |
4.28 |
1.21 |
10.7 |
2.9-3.78 |
|
I-caproico |
5.18 |
1.12 |
12.95 |
1.89-2.64 |
|
N-caproico |
5.81 |
1.28 |
14.52 |
4.95-6.3 |
|
eptanoico |
7.46 |
.. |
18.65 |
.. |
Nella Tab. 2, vengono riportati i valori medi degli altri parametri considerati: la selettività, la risoluzione dei picchi e il fattore di capacità. Tali parametri sono stati ottenuti per i tre livelli di concentrazione considerati. Nella tabella i parametri di selettività e risoluzione sono da considerarsi relativi al picco dell’acido sulla linea corrispondente e quello successivo.
Si può osservare dalla colonna relativa alla risoluzione, che per tutti i componenti la separazione è stata netta, tranne che tra acido propionico e I-butirrico, che comunque risulta dell’ordine del 98%. Dalla colonna relativa alla selettività si può notare che la capacità di risolvere i singoli picchi è stata buona.
Conclusioni
La metodica e le condizioni scelte hanno mostrato valori degli indici di controllo della qualità buoni o comunque accettabili per le analisi di controllo del processo anaerobico. In particolare si sono ottenuti buoni livelli di precisione, di risoluzione e di selettività, ottimi livelli di stabilità del fattore di risposta e valori accettabili in termini di accuratezza, per le esigenze di controllo del processo di digestione anaerobica. Il metodo inoltre presenta il vantaggio di un veloce e semplice pretrattamento del campione (solamente centrifugazione e filtrazione) e non necessita di metodi di estrazione o derivatizzazione che richiederebbero tempi lunghi che non aiuterebbero a prevenire brusche cadute di pH del processo di digestione anaerobica di reflui velocemente biodegradabili. Infine, un aspetto secondario, ma importante dal punto di vista gestionale quando vi siano diversi campioni da analizzare, è rappresentato dalla breve durata dell’analisi, dell’ordine dei 10 minuti primi.
Ringraziamenti
Si ringrazia il dottor Emanuele Sbaffi per la preziosa collaborazione.
Bibliografia
AA.VV. (1995). Standard Methods for the examination of water and wastewater. APHA, 19a ed.
Ahring B. K., Sandberg M. and Angelidaki I.,
(1999). Volatile fatty acids as indicators of process imbalance in anaerobic
digestion. Appl. Microbiol. Biotechnol.,
42, 559-565
Attal A., Ehlinger E., Audic J.M. and Faup G.M.
(1988). pH inhibition mechanisms of acetogenic, acetoclastic and
hydrogenophilic population. Paper presented at V Int. Symp. on Anaerobic
Digestion, Bologna (Italy), 22-26 May 1988.
Bottari E. e Liberti A. (1983). Analisi chimica quantitativa. Univ. Studi di Roma.
Calaresu G. (1994).
Guida all’applicazione delle Buone Pratiche di Laboratorio. SEF, Milano.
Clark and Speece (1970). pH tollerance of anaerobic digestion. Proceeding of V int. WPCF conference.
Jeris J. and Kugelman J. (1973). Anaerobic
digestion. Adv. Chem., 107.
Johnson E.L. and Stevenson R. (1978). Basic Liquid Chromatography. Varian.
Kodukula, Prasckam and Antonisien (1988). Role of pH in biological wastewater
treatment processes. In “Phisiological models in microbiology”, 1, 6, 113-135.
CRC Press, Boca Raton, USA.
Malaspina F., Cellamare
C. M, Stante L. and Tilche A. (1996). Anaerobic treatment of cheese whey with a
downflow–upflow hybrid reactor. Bioresource
Technology, 55, 131–139.
Malaspina F., Stante L., Cellamare C. M. and Tilche A. (1995) Cheese Whey and Cheese Factory Wastewater Treatment with a Biological Anaerobic–Aerobic Process. Water Science and Technology, 32, 12, 59–72.
Mentasti E. e Saini G.
(1990). Analisi chimica cromatografica. Piccin, Padova.
Pind F. P., Angelidaki I.and Ahring B. K.
(1999) The use of VFA measurements as process indicators in anaerobic reactors
treating manure. Vol. 1, oral presentation of " II International symposium
on anaerobic digestion of solid waste", 41-44, Barcelona, 15-17 June,
1999.
Rozzi A. (1984). Physico-chemical equilibrium in
anaerobic digestion. In “Biomethane use and production”, Pichen et al,
Turret-Wheatland ltd, Rickmansworth, UK.
Zehnder A.J. B. (1988). Biological of anaerobic microorganisms. John Wiley and Sons, New York.